Last October, the 3rd Aseptic BioPharma Processing Summit drew in attendees from all over the pharmaceuticals industry to Vienna for two days of presentations by expert speakers from across the pharmaceuticals industry, panel discussions and plenty of networking breaks.
Solution providers, researchers, drug manufacturers and industry experts from across the aseptic processing industry came together in Vienna to navigate the latest developments in aseptic processing, quality risk management, robotics and automation, microbial monitoring, automated visual inspection and more.
The conference attendees also discussed topics such as fill-finish tech transfer challenges, aseptic process simulation (APS) validation and considerations, barriers technologies, lyophilization, innovative models for aseptic manufacturing, sealing and packaging technologies.
This article will provide a session recap for those who didn’t get the chance to attend and serve as a reminder to those who attended.
Heart Beats of Aseptic Processing In Pharmaceutical Industry
Varadharaj Vijayakumar, Senior SME-Aseptic Fill Finish at WuXi Biologics
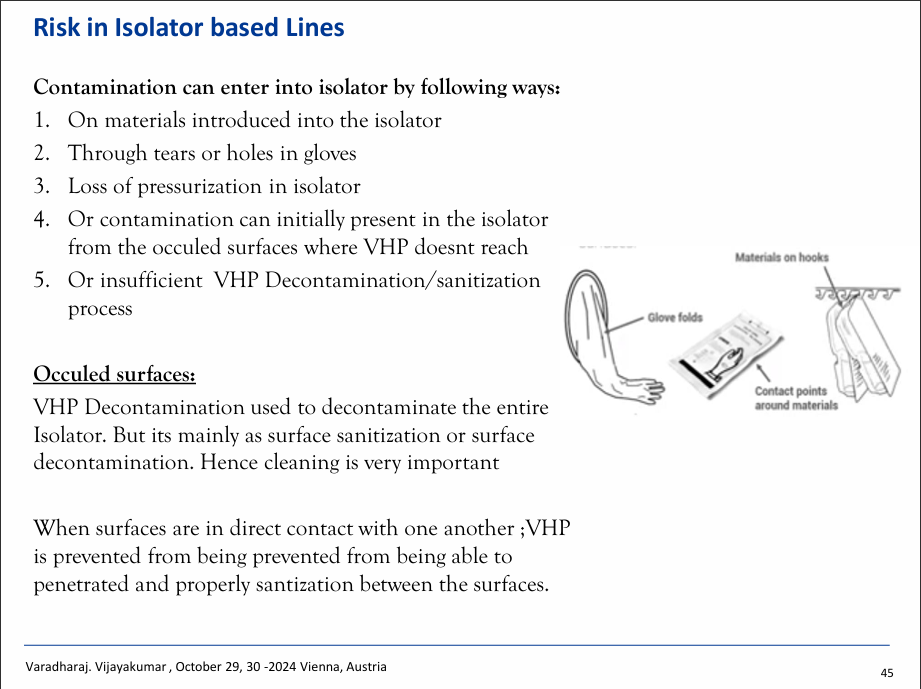
Varad discussed the four different types of inspection conducted by the FDA: pre-approval inspection, routine inspection, compliance follow-up inspection, and “for cause” inspection. He shared how to reduce loss in the filtration stage for isolator-based filling lines by reducing the length and inner diameter of the tubings and using filters with smaller surface areas. Losses can also be reduced by utilising proper fill volume settings, trained operators and clear process and procedural controls. Sterility assurance can be prioritised by taking measures such as being cautious of all PPM, using E beams for external decontamination, and using a no-touch transfer system to introduce pre-sterilized material into a Grade A cleanroom. Other measures such as using multiple layers and removal of packaging may reduce the risk of contamination, improving operator training, the usage of Rapid Transfer (RT) containers and the visual inspections of gloves pre- and post-use can also aid sterility assurance.
Challenges in Industrial-Scale Fill & Finish Operations for High-Concentration Proteins Fill & Finish Process
Jean-Rene Authelin, Senior Scientific Advisor Global CMC at Sanofi
High-concentration (50 to 300mg) protein self-administered therapeutic doses are used for patient convenience but present processing difficulties for manufacturing. For the product to be able to be used in syringes or autoinjectors the viscosity must be kept below 20-25cP at room temperature as increased viscosity increases injection time but the use of a higher concentration of proteins increases this value. CFD simulations show the mixing time during the fill-finish process is 10 to 100 times larger than expected, the filtration rate is inversely proportional to the viscosity requiring larger filters and filling is made more difficult to variable viscosity based on temperatures, prejudicing filling robustness.
Fill finish operations for high-potent products – with a case study for an ADC facility
Lukas Munzinger, Global Product Manager at Syntegon Technology GmbH
Lukas began his presentation by recapping drug product trends, explaining how 25% of injectable biologics are cell & gene therapies, the future big role that isolators will play as manufacturing of high-potent drugs will become more important, and how oncology drugs have the second highest growth and will need equipment supporting their manufacturing challenges. For working with potent and high-potent products there are 2 important values, Occupational Exposure Limit (OEL) which is the limit an operator can be exposed to a certain product on average during one shift (8 hours) without affecting health and Occupational Exposure Band (OEB). which groups several OELs together. OEB classification varies according to several national standards as well as classifications within different companies. Choice of barrier system and additionally needed measures are selected according to OEB class, GMP vs. EHS (environment, health, safety) requirements and the specific needs for high-potent manufacturing in equipment design. Lukas shared a case study of the fill-finish process of ADC products, highlighting the importance of thoroughly defining and reviewing processes and close collaboration with equipment suppliers and/or engineering companies.
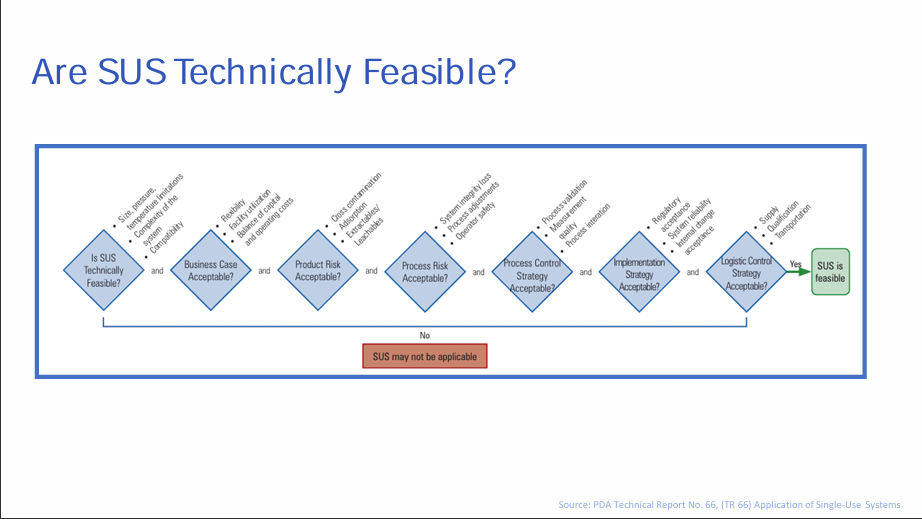
The Challenge of Single Use Systems
Eva Ulied, Production Lead and Alexandra Gonzalez, Quality Lead, at Boehringer Ingelheim
The pair from Boehringer began their presentation with the challenges of implementing single-use systems which require specialised technical expertise for installation, operation, and maintenance which is difficult to acquire. Extensive validation and qualification are crucial for ensuring the performance and reliability of single-use systems. This process can be time-consuming and costly, often requiring specific expertise and equipment. Integrating single-use systems with existing infrastructure and equipment can be complex, requiring careful planning, design, and validation to ensure seamless operation and compatibility. Proper training is crucial, ensuring operators are familiar with single-use technology can be done by enforcing a general SOP for handling SUS with detailed work instructions covering all steps and potential errors with a graphical overview of materials and connections. Regarding Quality Assurance requirements in Annex I, single-use systems face regulatory scrutiny as agencies like the FDA and EMA have specific regulations regarding the manufacturing, validation, and documentation of single-use components.
Ensuring Product Sterility in Processing Frozen Sterile Bulk Drug Product in Single-use Bag Assembly
Yuan-An Liu, Associate Director CMC at BioNTech SE
Yuan-An addressed the challenges in processing frozen sterile bulk drug products, discussing how the size of mRNA lipoplexes used in drug delivery is larger than the pore size of sterilising filters, manufacturing of RNA-LPX and fill-finish in one facility is not possible and the restricted stability of mRNA. Existing control strategies involve the use of single-use assembly (SUA) where the product is formulated, delivered to a secondary manufacturing site and fill-finish is carried out. This requires various measures to ensure product sterility such as integrity testing of SUA at vendor release, qualification of SUA components under processing conditions, sterility testing at the release of sterile bulk and secondary packaging. Yuan-An also discussed how developing a contamination control strategy should include 3 stages: risk assessment, risk mitigation, and risk reassessment. A qualification study should be carried out on the risk mitigation stage to prove that the SUA is integral enough to prevent microbial penetration even under worst-case process conditions and challenging bacterial soiling.
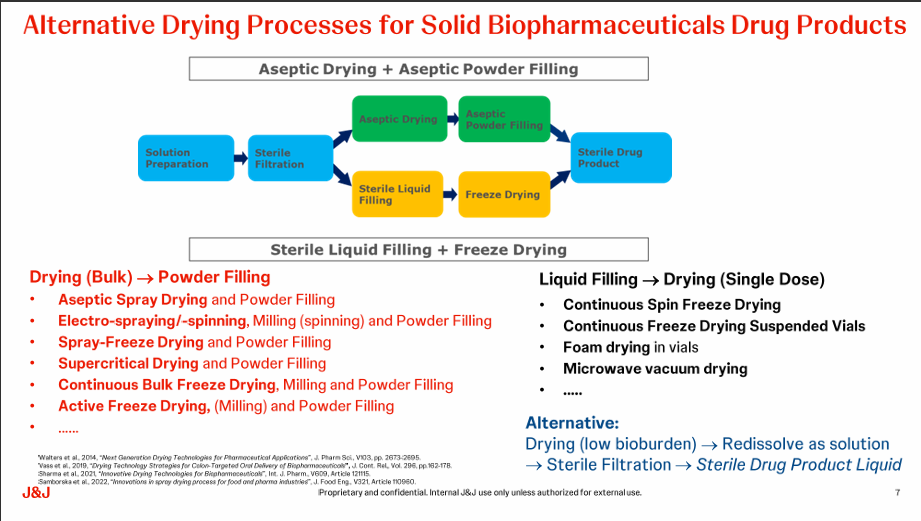
Current Trends in Aseptic Drying Technologies
Sune Klint Andersen, Scientific Associate Director at Johnson&Johnson Innovative Medicine
Sune shared the challenges faced in the freeze-drying process such as the risk of burying quality that can conflict with FDA/EMA guidelines, lack of flexibility and its time-consuming nature. He discussed two aseptic drying technologies – thin-film freeze drying and spray freeze drying. Both have issues with scaling up as well as freezing and dehydration stresses but are well-suited for heat-sensitive pharmaceuticals. Aseptic spray drying technology is gaining interest for vaccines and highly concentrated mAb formulations but has limited capacity and expertise, however, smaller aseptic spray dryers are becoming available from several manufacturers. Aseptic freeze-drying technologies are available for spray freeze-drying and continuous vial freeze-drying suppliers but have limited capacity.
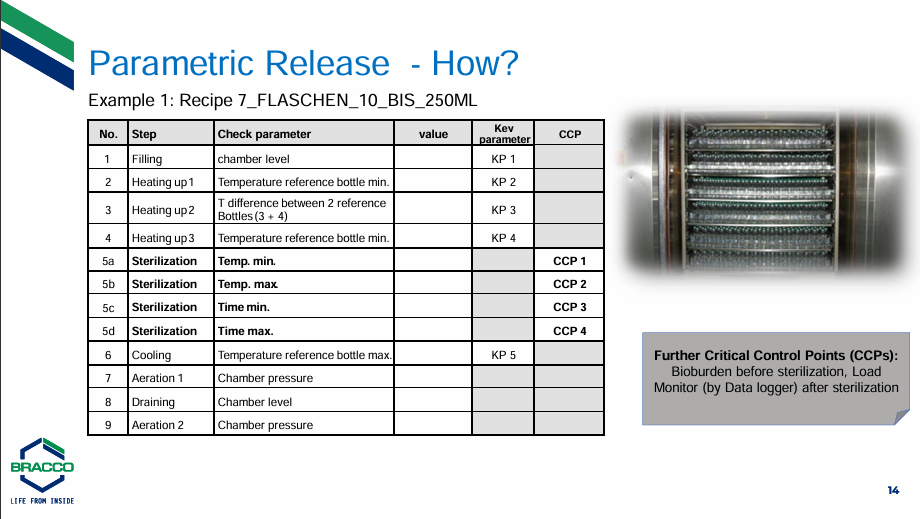
Parametric Release – a smart alternative for terminally sterilized products
Stephan Heck, Director of Quality at BIPSO GmbH
Stephan began his talk by speaking about the use of contrast media for X-rays and MRIs and how the iodine contrast media used in X-rays and the gadolinium contrast media used by MRIs have a parenteral application, being expelled via glomerular filtration through the kidneys. He recapped the regulatory background of and preconditions for Parametric Release drugs, as it can only apply to products terminally sterilised in their final containers. Key aspects of Parametric Release include an integrated approach to secure sterility for the next level of microbiological risk mitigation and avoidance of false positive results. However, attention must be given to the autoclave load pattern, reprocessing procedure, including Parametric Release CCPs/CPs into your CPV/PQR Program and creating a Parametric Release specific test instruction.
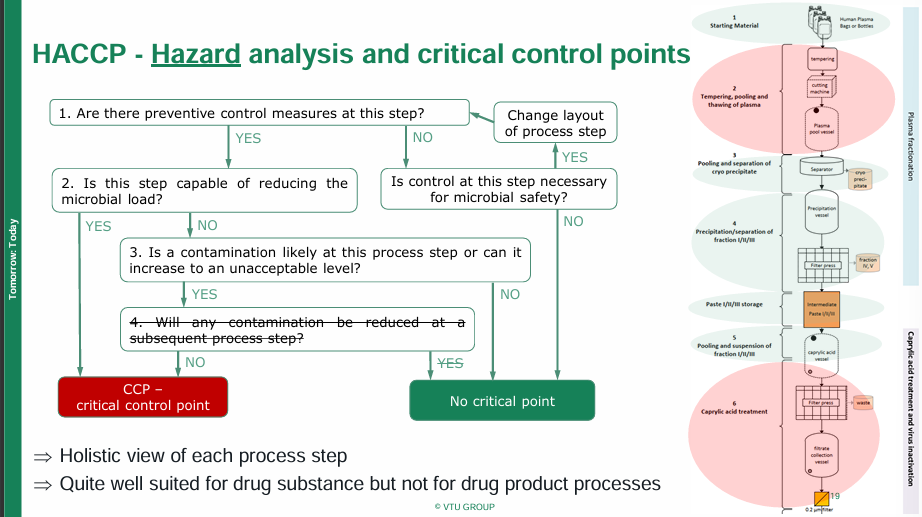
Annex 1 Contamination Control Strategy
Cornelia Haas, Manufacturing Science and Technology Team Lead at VTU Engineering
Cornelia addressed the implementation of a contamination control strategy that adheres to Annex I guidelines in her keynote speech. The creation of a main document that includes the pharmaceutical quality system, facility, utilities, equipment, materials, personnel, and process with general descriptions, the Annex I requirement and the level of detail required is good practice. A contamination risk assessment for every process must be undertaken as well for each product separately vs. generic that is interlocking with the main document. The risk analysis should include failure mode and effect analysis, hazard analysis and critical control points and a preliminary hazard analysis. Risk should be evaluated by calculating the risk priority number and comparing it against the risk matrix, where it can be classified as an acceptable residual risk, justified risk acceptance/defined mitigation action or requiring additional risk control.
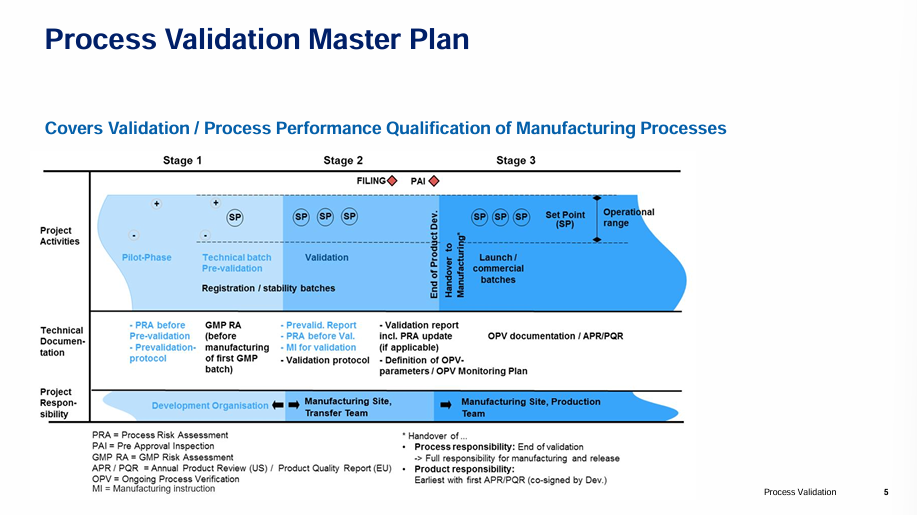
Process Validation Sterile Drug Products: strategy, execution and maintaining the validated state
Sandra Hedtke, Senior Technical Tranfer Lead at Novartis
Sandra introduced how to create a site validation master plan, covering topics such as work packages and meeting international and internal regulations, all included in an overarching document referencing individual VMPs and Guidance SOPs of the individual systems. She also discussed how to create a Process Validation Concept, by first tackling Process Design and defining the commercial manufacturing process, then Process Qualification/Validation where it is evaluated to assess whether the process is capable of reproducible commercial manufacturing before finally, implementing Ongoing Process Verification (OPV) for ongoing assurance that the process remains in a state of control. Revalidation due to changes, the scope of any necessary activity will be assessed within the framework of change.
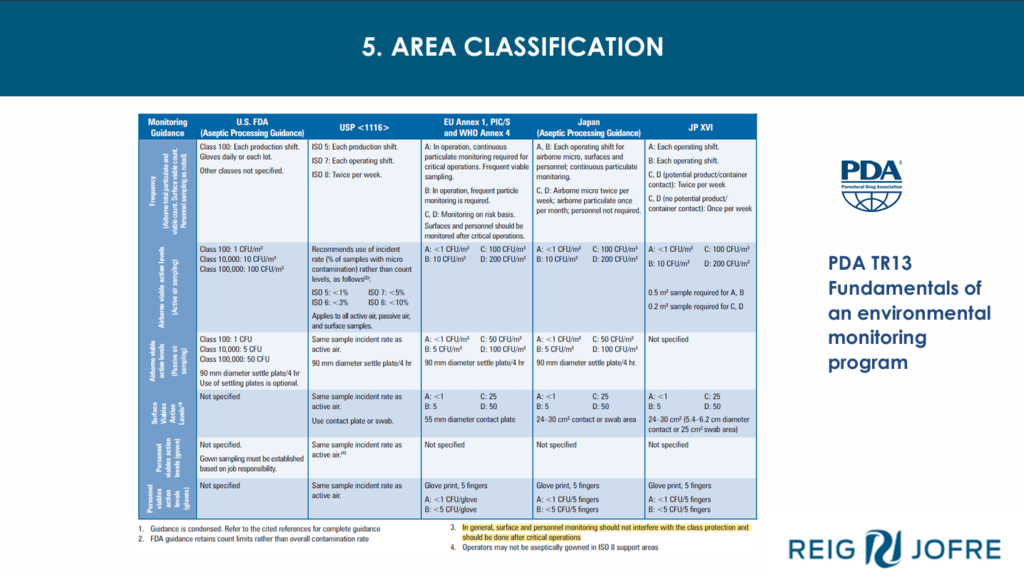
Environmental Monitoring, Qualification and Routine Environmental Monitoring in Classified Areas, RABs and Isolators
Beatriz Roqués, QA Aseptic Process Manager at Reig Jofre
Beatriz introduced the importance of environmental monitoring in qualification processes for aseptic processing. She discussed microbial environmental monitoring such as trend reports for counting and flora and results reading, and what is involved in Performance Qualification of environmental monitoring (EMPQ). EMPQ involves evaluating the main flora, in-house and objectionable microorganisms, alert limits definition, deviations, data management, reports and evaluation, performing as built, static and in operation phases (PQ), defining locations and frequencies and the design of the plant, rooms and equipment. Area classification categorises clean rooms into assigned levels of clean air, measured against an acceptance criteria of grade A (highest) to grade D (lowest) and based on total particulate concentration for classification and monitoring and maximum permitted microbial contamination during qualification, maximum action limits for viable particle contamination. The first step to assessing risk is to decide its severity and its probability. The class of the risk analyzed is obtained from the values of these parameters and final locations must be justified by RA. Using automated systems with the aid of barcode readers and software for EMPQ cuts the overall time by half. Beatrix concluded her presentation with excerpts from the Annex I guidelines for the points she covered in her talk.
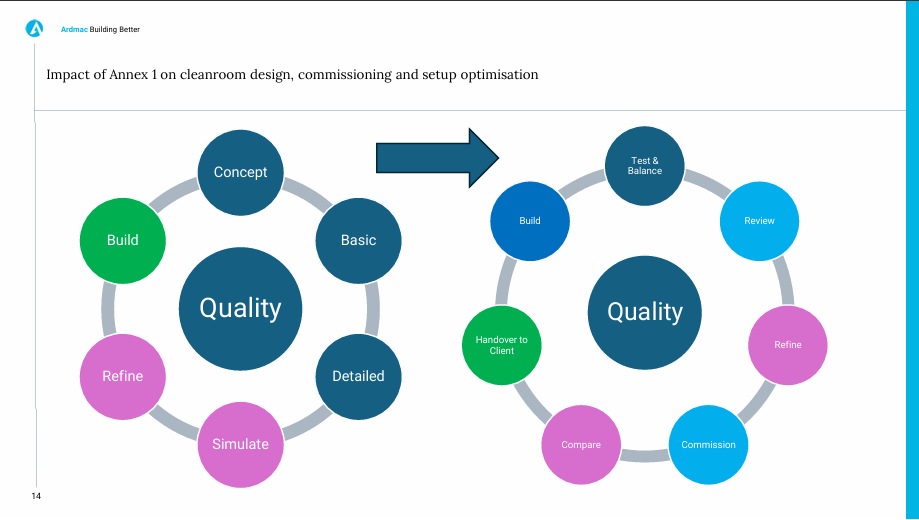
Impact of Annex 1 on cleanroom design, commissioning and setup optimisation
John Carey, Head of Engineering at Ardmac Group
John used his experience as a Head of Engineering to discuss how contamination control can be approached as early as the design stage. The designer needs to be educated on how the end user envisages the process working and work with the Quality Department to account for process flows and contamination sources in their design. Using zoning in the design stage to group contamination risks and isolation and barrier technology to target known contamination sources. Quality Risk Management is also accounted for in the design stage, using flexibility to change according to data gained from risk assessments. The Turnover Process is essential to ensuring the design has been implemented as intended, so we know what the cleanroom is capable of in the future. John ended his speech listing the key takeaways, the importance of early engagement and integration between all parties, designers engaging with the Quality Department, using flexibility to design for refinement and reviewing and improving after commissioning.
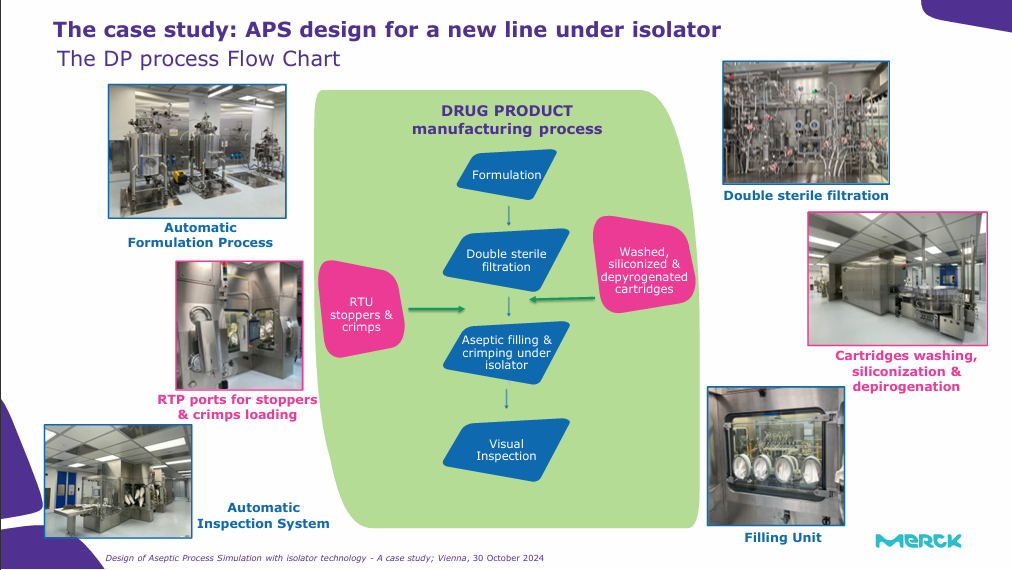
Design of Aseptic Process Simulation with isolator technology – a case study
Sara De Pinto, Aseptic Operations Expert at Merck Group
Sara discussed the Aseptic Process Simulation (APS) in light of the new Annex 1 guidelines. APS is a key element of the overall Contamination Control Strategy, the object of systematic attention from regulatory assessors during approval and periodic revision processes for products and manufacturing sites/lines. Quality Risk Management (QRM) from Ref. ICH Q9 is applied to Aseptic Process Simulation, with an increased focus on worst-case conditions identification and on appropriate documentation level for justifications and rationales, risk assessments. Sara introduced a case study for an APS design for a new line under an isolator which employed the strategy of a risk-based approach, worst-case conditions identification and continued revision of the APS design based on manufacturing experience. Sara concluded that the adoption of QRM principles is key to designing a robust simulation strategy, able to properly address all potential risks for the aseptic process and based on the proper selection of worst-case conditions to be challenged and good documentation of scientific and technical rationales is integral part of an overall properly set APS design.
Use of AI in automation of reading Environmental Monitoring plates
Andrew Gravett, Principal Scientist, Microbiology at AstraZeneca
Andrew introduced the Automated Plate Assessment System (APAS), an automated agar plate reader that uses a camera system and an Artificial Intelligence (AI) model to count and sort plates. The APAS system has already been approved for use in clinical spaces by the FDA and in Europe and Australia. AstraZeneca partnered with creators Clever Culture Systems to develop the system for the pharma industry as APAS is able to read and verify 30,000 EM agar plates and it resolves data integrity challenges. APAS processes and categorises ~200 plates/hour, only requiring technicians’ time to check plates with growth or processing errors and automatically transferring data to LIMS system. The AI model is trained and tested by qualified microbiologists and allows all this variability to be managed. AstraZeneca carried out secondary validation, stage 1 established the expected performance and showed APAS only missed growths on 15 out of 2557 compared to 27 by the human manual count. Stage 2 established real in-use performance and APAS missed 11 plates with growth out of 7424 compared to 7 from the manual counts. Counting is impacted by areas of growth confluence where it is difficult to accurately count and can also be subjective between operators, the most important feature of APAS is single colony detection and the ability to sort Growth from No Growth. The data shows equivalent false negatives to the manual method and around 7% false positives. Andrew concluded by assessing the potential risks and future considerations such as potential revalidation, expansion to include different plate sizes and consideration for image retention.
Validation of Biotech API Products: ANNEX 1 Interpretation Requires Clarification
Nigel Cryer, Managing Director at Bluciela Lifesciences
Nigel began his presentation on how to apply and design Annex I into the API/bulk process by undertaking a process risk assessment. He discusses regulatory inspection preparation, focusing on the validation of biotech API products per EU GMP Annex 1, emphasising sterility assurance, bioburden reduction, risk assessment, and compliance with data integrity and ICH Q7 guidelines. He concluded that complying with the EU GMP Annex 1 requirements for the manufacture of sterile medicinal products is crucial for pharmaceutical manufacturers, understanding the guidelines, undertaking risk assessments and adopting a risk-based approach helps ensure compliance.
Microbial Identification in the Pharmaceutical Facility: An EMPQ Approach
Christian Scheuermann, Global Technical Services Manager at Charles River Laboratories
Christian introduced his topic with a recap of the GMP Annex 1 guidelines regarding prevention – tracking and trending, environmental monitoring, aseptic process simulation, bioburden and disinfection. he highlighted the importance of microbial identification as the species level ID is providing all information that is relevant to keep the manufacturing environment in a state of control. Accurate identification in EMPQ from various sampling points creates a surveillance system, indicating where microbes reside and how they might potentially spread, how far they will resist cleaning procedures, whether they produce endotoxins or toxins in general, the risk to the facility, products and staff, how to improve sterility assurance and how to create cost avoidance savings that are due to inefficient CAPA.
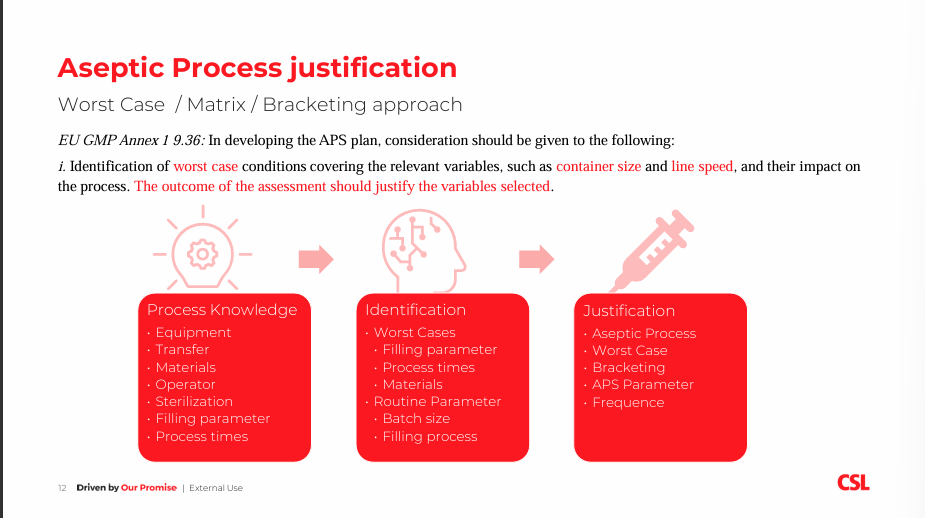
Annex 1 requirements for APS and Operator Qualification with insight from Swissmedic Q&A
Gesine Kaiser, Site Leader Sterility Assurance at CSL Behring
Annex I guidelines advise the use of Aseptic Process Simulation in order to verify the capability of the process to assure product sterility. Gesine discussed the aseptic process justification by using worst-case conditions that would pose the greatest chance of process or product failure when compared with ideal conditions. She described the Swissmedic interpretation of GMP Annex I regarding the worst case/matrix approach for APS, interventions and operator qualification. She concluded that APS must be designed with a basis of solid manufacturing process understanding, assessed and justified parameters, a rationalised approach, and a risk assessment of interventions carried out based on process understanding and design. Gesine also covered operator qualification and how adequate training and re-training of employees working in grade B and A is required including theoretical knowledge transmission and practical training based on activities of the individual employee. Training effectiveness must be assessed, as well as operators requiring initial qualification and annual re-qualification through participation at APS.
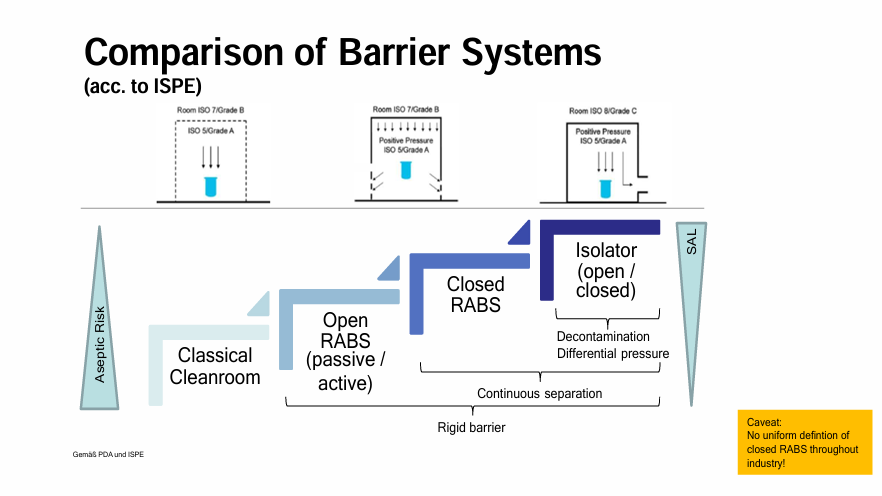
New Cleanroom and new barrier system requirements from Annex 1
Doris Lacej, Head of Parenteral Solutions Department at Profarma
Doris began her keynote speech with a recap of the Annex 1 revision regarding isolators introducing the concept of open isolators. Isolators per se are no longer perceived as the ultimate response to aseptic challenges, open isolators change the focus from the aseptic process to its environment. Annex I changes the minimum requirements for room classification from Class D to Class C, requiring new concepts for established clean rooms. The new EU GMP Annex I tells us barrier systems are state-of-the-art and are preferred against classical cleanroom with no preference between RABS or isolators. Doris compared RABS and Isolators, discussing their characteristics and weighing up their benefits and challenges, and their costs and installation.
ABPP24 was supported by a wide range of companies who brought their teams to our exhibition hall and Innovatrix would like to thank our sponsors again.
If you want to attend our next leading event focused on advancing the standards and practices in sterile pharmaceutical production and have the opportunity to hear presentations like these and many more, join us for our fourth edition of this summit. Explore the latest trends, technologies, and regulatory requirements in aseptic manufacturing, meet with solution providers and hear talks from industry leaders, attend the 4th Aseptic BioPharma Processing Summit taking place in Vienna, Austria on October 7-8, 2025.
For more information, visit our website or email us at info@innovatrix.eu for the event agenda. Visit our LinkedIn to stay up to date on our latest speaker announcements and event news.
This website uses cookies so that we can provide you with the best user experience possible. Cookie information is stored in your browser and performs functions such as recognising you when you return to our website and helping our team to understand which sections of the website you find most interesting and useful.
Strictly Necessary Cookies
Strictly Necessary Cookie should be enabled at all times so that we can save your preferences for cookie settings.
If you disable this cookie, we will not be able to save your preferences. This means that every time you visit this website you will need to enable or disable cookies again.